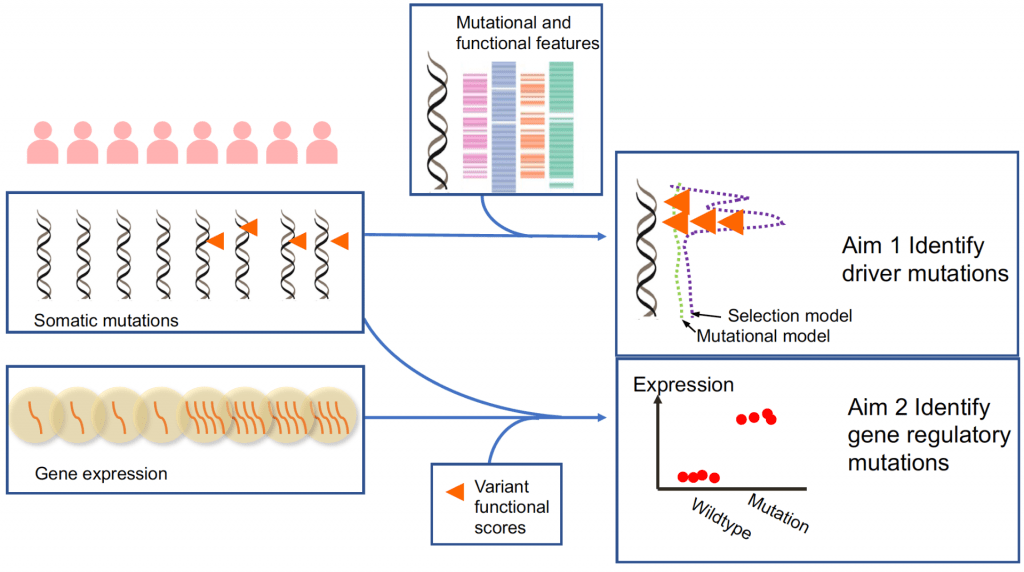
One major goal of cancer genome sequencing is to identify driver mutations and understand how they contribute to tumorigenesis. The analysis of somatic mutations in protein-coding regions, specifically, has yielded highly fruitful results, highlighting novel and potentially impactful genetic mechanisms and therapeutic strategies for treating cancer. The vast majority of somatic mutations, however, occur in noncoding regions and their roles in cancer are not well understood. There are multiple difficulties in evaluating the roles of noncoding mutations. For example, the complex background genetic changes in cancer often obscure functional signals, the lack of functional annotation for noncoding regions makes interpretation of results difficult and the paucity of strong selection signals compared to mutations in coding regions requires more powerful tools.
Several strategies have been utilized to identify and investigate somatic noncoding mutations that serve as drivers for cancer. Researchers have developed methods based on the recurrence of mutations in regulatory elements, the clustering pattern of mutations, or the enrichment of mutations with functional annotations. To evaluate if noncoding somatic mutations have regulatory impacts, a few studies have tried to perform association tests between somatic mutation status and nearest gene expression level. Several drawbacks of current methods hamper the power for making more discoveries. Many lack explicit statistical models to distinguish selection signals from background mutational process. Current methods are also not flexible enough to integrate the context-dependent effects of noncoding mutations into their analysis. Finally, recurrent noncoding mutations are rare and standard association methods that focus on individual loci are underpowered.
In this project, we aim to develop statistical methods to overcome these drawbacks and provide a more comprehensive analysis of noncoding somatic mutations in cancer. We will leverage our expertise in modeling positive selection and mapping of regulatory variants to tackle this problem. In collaboration with the Center for Quantitative Biology (CQB) Data Analytics Core and Single Cell Genomics Core, we will then apply these methods on large-scale genomic datasets of tumor samples and perform computational and primary experimental validation for our findings.